 S A N A الوكالة العربية السورية للأنباء
S A N A الوكالة العربية السورية للأنباء

اللاذقية-سانا
أعلن أستاذ في جامعة رايس الامريكية غانغ باو مؤخرا نجاح عملية تعديل جينية لإصلاح 20 إلى 40 بالمئة من الخلايا الجذعية المأخوذة من الدم المحيطي لمرضى فقر الدم المنجلي ما يشكل أملا جديدا لعلاجهم.
وجاء إعلانه في الاجتماع السنوي للجمعية الأمريكية لتقدم العلوم بعد سلسلة دراسات مع كلية بايلور للطب ومشفى تكساس للأطفال وجامعة ستانفورد لإيجاد علاج لاضطرابات الخلايا المنجلية أو فقر الدم المنجلي وهو مرض وراثي يحدث نتيجة وجود طفرة تجعل خلايا الدم الحمراء على شكل هلال وتعوق تدفق الدم في الأطراف والأعضاء.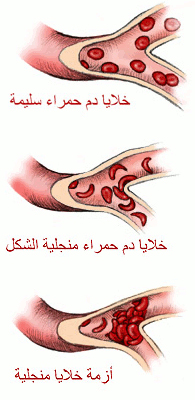
وقال باو “اضطراب الخلايا المنجلية يحدث بسبب طفرة واحدة لذلك نعمل على تصحيح هذه الطفرة للحصول على خلايا دم سليمة لكن لا نعرف إن كان إصلاح ما يصل إلى 40 بالمئة من الخلايا كافيا لعلاج المريض هذا شيء نأمل أن نكتشفه من التجارب السريرية”.
وحتى الوصول إلى علاج نهائي لفقر الدم المنجلي يوصي اختصاصي الدم والأورام عند الاطفال الدكتور عبدالمنعم غانم بعدم زواج الاقارب لأن المرض وراثي وإجراء رحلان خضاب لكل المقبلين على الارتباط لكشف ما إذا كان الطرفان يحملان المرض.
وأوضح الدكتور غانم أن اكتشاف المرض غالبا ما يكون خلال العامين الأولين من عمر الطفل بعد شكوى مستمرة من حرارة وتورم في مفاصل القدمين واليدين مع شحوب الوجه الأمر الذي يثبت بالتشخيص وفحوص رحلان الخضاب.
وفي حال تأكد الإصابة يوصي الاختصاصي الأهل بالانتباه لحالة الإماهة لطفلهم المريض ومعالجة الانتان فور حدوث أي ترفع حروري بسب ضعف مناعته واعطاءه حمض الفوليك يوميا مدى الحياة واستكمال اللقاحات المدرجة في البرنامج الوطني.
ويشدد الدكتور غانم على ضرورة مراجعة الطبيب فورا عند حدوث أي نوبة ألم في الأطراف أو البطن أو الصدر للمريض منعا من تفاقم الحالة مبينا ان مريض فقر الدم المنجلي لا يتطلب نقل دم متكررا كما في التلاسيميا.
ونبه الدكتور غانم إلى ضرورة التغذية الجيدة للطفل ومراقبة حالته الصحية باستمرار عبر الفحوص السريرية والمخبرية والشعاعية لتجنب اختلاطات المرض الخطرة.
بشرى سليمان